Eine der häufigsten Erbkrankheiten
Zystische Fibrose stellt eine der häufigsten Erbkrankheiten in der westlichen Welt mit einer Inzidenz von 1:2500 Geburten dar. Sie wird autosomal-rezessiv vererbt bei einer Trägerhäufigkeit von 1:20 – 1:25 Personen.
Εs handelt sich um eine schwere Erkrankung, die viele Organe, wie Atmungsorgane, Pankreas und Organe des Magen-Darm-Traktes betrifft. Bei Männern können einige Mutationen Unfruchtbarkeit verursachen (bilateraler Mangel an Samengang (CBAVD).
Mutationen von Zystischer Fibrose
Das für die Krankheit verantwortliche Gen wurde 1989 identifiziert und heißt Regulator des transmembranösen Transports von Ionen (cystic fibrosis trans membrane regulator, CFTR).
Bis heute sind mehr als 1600 Läsionen (Mutationen) im genetischen Material der Patienten mit zystischer Fibrose nachgewiesen, die meisten davor gelten als pathologisch und zeigen eine unterschiedliche geografische und demografische Frequenz.
Die häufigste Mutation in Griechenland mit einer Frequenz von 53,4% ist ΔF508. Sie gilt die Symptomatik betreffend als eine der schwersten Mutationen.
Die nächsthäufigsten Mutationen, die weitere 17,7% -der Bevölkerung in Griechenland umfassen, sind:
- 621+1G>T (5,7%)
- G542X (3,9%)
- N1303K (2,6%)
- 2789+5G>A (1,7%)
- 2183AA>G (1,4%)
- E822X (1,4%)
- R1158X (1%)
Die restlichen Mutationen der Krankheit haben eine Inzidenz von weniger als 1%.
Zystische Fibrose und Befruchtung
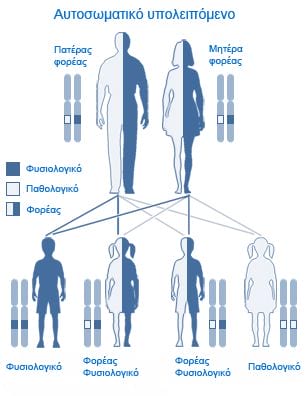
Jeder Mensch trägt zwei Kopien des CFTR-Gens in sich. Damit eine Erkrankung an zystischer Fibrose erfolgen kann, muss die Person die Mutationen in beiden Kopien des CFTR-Gens in sich tragen
sogenannt -homozygot– während jemand, der nur an einer Kopie des Gens eine Mutation trägt, als heterozygoter Träger gilt.
Damit ein Kind nicht mit zystischer Fibrose geboren wird, sollten entweder beide Elternteile keine Mutationen auf den Kopien der CFTR-Gens tragen oder falls eine Mutation vorhanden ist, sollte dies nur bei einem Elternteil der Fall sein. Wenn beide Elternteile Träger der Mutation sind, besteht eine 25%ige-Wahrscheinlichkeit, dass ein Kind mit zystischer Fibrose geboren wird.
Untersuchung der Zystischen Fibrose
Der einzige Weg zur Erkennung der Träger von zystischer Fibrose ist die molekulargenetische Untersuchung, d.h. der Nachweis von Mutationen im CFTR-Gen. Die Untersuchung für zystische Fibrose zur Erkennung von Trägern erfolgt:
- in Familienplanung-Fällen
- bei positiver Familienanamnese
- bei Darmechogenität im Ultraschall
- in der Unfruchtbarkeitsuntersuchung
Im Fall einer bevorstehenden Familienplanung und dem Wissen, dass, beide Elternteile Träger der Mutation sind, werden Informationen über den Embryo durch die Methoden der Präimplantations